1. Introduction: A Primer on Wilms Tumor (Nephroblastoma)
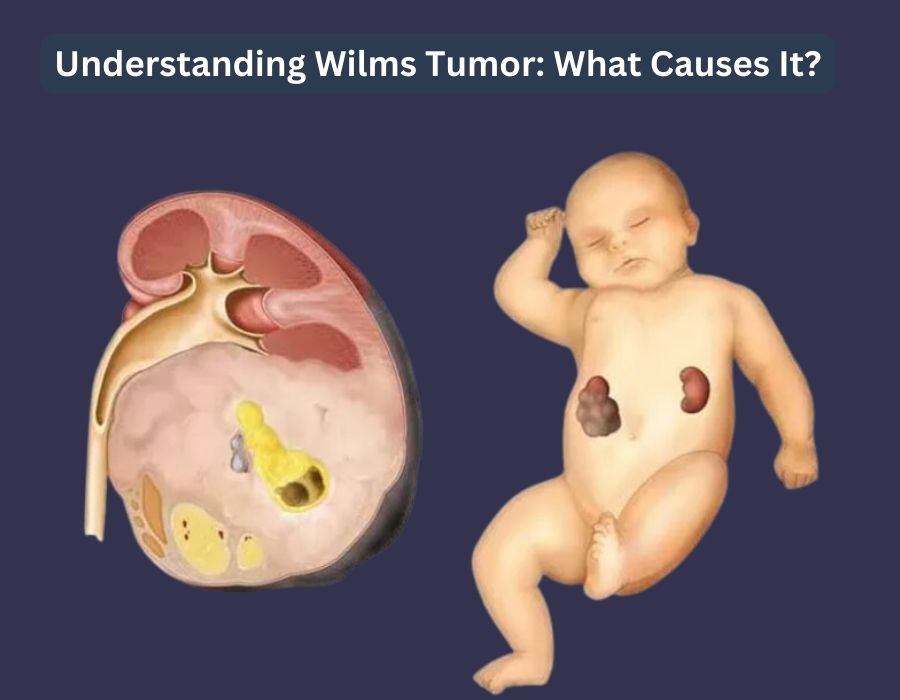
Wilms tumor, a form of kidney cancer that predominantly affects children, is one of the most significant topics in pediatric oncology due to its unique pathophysiology, high cure rates, and role as a model for modern cancer management. The disease is also referred to as nephroblastoma, a name that more accurately reflects its origin from embryonal kidney tissue. This dual nomenclature highlights a fundamental distinction between the disease’s historical identification and its modern pathological understanding, a detail of central importance in expert discourse. The name “Wilms tumor” honors Max Wilms, the surgeon who first provided a comprehensive pathological description of the disease in the early 20th century. However, the term “nephroblastoma” directly links the malignancy to its cellular origin—the metanephric blastema, the precursor tissue of the kidney—placing it within a precise developmental and cellular context.
Pathologically, Wilms tumor is an embryonal tumor that typically presents with a triphasic histological pattern, meaning it contains three distinct components: blastemal, stromal, and epithelial tissues. The proportions of these components can vary significantly from one tumor to another, resulting in a wide range of appearances under the microscope. The blastemal component is considered the least differentiated and most malignant, consisting of small, undifferentiated cells with high mitotic activity. The epithelial component forms primitive tubules and glomeruli, while the stromal component comprises differentiated cells such as smooth muscle, fibroblasts, and cartilage. The presence and relative abundance of these components, as well as the presence of anaplasia, are critical factors in determining the tumor’s prognosis and guiding treatment decisions.
2. Etiology and Pathogenesis: A Molecular and Syndromic Perspective
The development of Wilms tumor is intrinsically linked to a series of developmental and genetic anomalies that disrupt the normal maturation of kidney cells. The origin of the disease is believed to be from immature kidney tissues known as nephrogenic rests. These are abnormal groups of kidney cells that fail to mature properly during fetal development and can persist in one or both kidneys after birth. While nephrogenic rests themselves are non-cancerous, they are susceptible to further genetic changes that can trigger their transformation into a tumor. This developmental pathology is a key feature of Wilms tumor, explaining its occurrence primarily in young children whose kidneys are still undergoing development.
The molecular underpinnings of Wilms tumor involve alterations in a number of critical genes that regulate cell growth and division. The WT1 (Wilms Tumour 1) gene, located on the short arm of chromosome 11, was the first gene identified in association with the disease. The WT1 gene encodes a transcription factor essential for the normal development of the urogenital system. In the context of Wilms tumor, it primarily acts as a tumor-suppressor gene, and its inactivation leads to the unchecked proliferation of cells and tumor development. It is a notable biological phenomenon that the same WT1 gene can behave as an oncogene in other malignancies, such as leukemia and lung cancer. This apparent contradiction highlights a fundamental principle in molecular biology: a gene’s function as a proto-oncogene or a tumor suppressor is not static but is profoundly influenced by its specific cellular environment and the signaling pathways in which it is involved.
While WT1 is a critical factor, mutations in this gene are found in only 5% to 10% of Wilms tumor cases, indicating a complex and multifactorial etiology. Other gene variants frequently associated with the disease include CTNNB1 (on chromosome 3) and AMER1 (on the X chromosome), both of which provide instructions for proteins that regulate gene activity and cell proliferation. A separate but equally important mechanism involves genomic imprinting on the short arm of chromosome 11, which contains the IGF2 and H19 genes. Genomic imprinting is a phenomenon where a gene’s activity is determined by whether it was inherited from the mother or the father. In some cases of Wilms tumor, an abnormality in this process leads to a loss of activity in the H19 gene, which normally restrains cell growth, and an increase in the activity of the IGF2 gene, which promotes it. The combined effect of these changes is uncontrolled cell growth and tumor formation. This genetic predisposition also manifests in a higher incidence of Wilms tumor among individuals with certain congenital syndromes. These syndromes demonstrate how pre-existing genetic conditions can create the cellular environment necessary for the development of Wilms tumor, making affected individuals highly susceptible to the disease.
Table 1: Common Genetic Syndromes Associated with Wilms Tumor
| Syndrome | Associated Gene(s) | Corresponding Wilms Tumor Risk |
| WAGR Syndrome | WT1, PAX6 (on 11p13) | 50% risk |
| Denys-Drash Syndrome (DDS) | WT1 | >90% risk |
| Frasier Syndrome | WT1 (intron 9) | Low but elevated risk, exact risk unknown |
| Beckwith-Wiedemann Syndrome (BWS) | IGF2, H19 (on 11p15) | Associated risk, exact risk unknown |
WAGR syndrome is characterized by a chromosomal deletion at 11p13 and is defined by the presence of Wilms tumor, aniridia (partial or complete absence of the iris), genitourinary anomalies, and developmental delays. Individuals with this syndrome have a 50% chance of developing Wilms tumor. Denys-Drash syndrome, caused by heterozygous mutations in the WT1 gene, is a more severe condition with a strikingly high risk of Wilms tumor, affecting more than 90% of individuals. Frasier syndrome is another WT1-related condition that carries a slightly increased, though overall low, risk of Wilms tumor but is associated with a high risk of gonadoblastoma in males. Lastly, Beckwith-Wiedemann syndrome, linked to the genomic imprinting of the IGF2 and H19 genes, is characterized by overgrowth, large organs, and hemihypertrophy (abnormally large growth on one side of the body). The identification of these syndromes provides invaluable insight into the developmental and genetic origins of Wilms tumor and underscores the importance of a comprehensive genetic evaluation.
3. Epidemiology and Risk Factors
Wilms tumor is the most common kidney cancer in children, yet it remains a rare disease. In the United States, approximately 500 to 650 new cases are diagnosed annually, a number that has remained stable for many years. This translates to an age-adjusted incidence rate of about 5.7 cases per million children. In Europe and North America, the global incidence is estimated at 1 in 10,000 children. Despite its low incidence, the disease is the subject of extensive, collaborative research efforts worldwide. This is not a contradiction but a testament to its status as a model tumor for modern pediatric oncology. The success in treating Wilms tumor, with its well-defined genetic basis and high cure rates, allows the field to focus on refining treatment protocols to reduce long-term side effects, a strategic goal that can then be applied to other childhood cancers.
The disease most frequently affects young children, with the average age of diagnosis being 3 to 4 years old. It becomes progressively less common as children grow older and is exceedingly rare in adults. Epidemiological studies have identified several demographic and racial trends. Wilms tumor is slightly more common in females than in males. In North America and Europe, the risk is slightly higher for Black children compared to White children, while Asian American children have a lower risk. Other risk factors include a family history of Wilms tumor, as well as the presence of certain congenital conditions like aniridia, hemihypertrophy, and genitourinary tract problems, which are frequently part of the syndromes described in the previous section. A mother’s exposure to pesticides during pregnancy has also been suggested as a possible risk factor.
4. Diagnosis, Staging, and Histological Classification
The diagnostic process for Wilms tumor is a critical, multi-step procedure aimed at confirming the presence of a renal mass, differentiating it from other kidney tumors, and determining the extent of its spread. The process often begins when a physician feels a mass in the child’s abdomen during a routine physical examination.
4.1. Diagnostic Modalities
A range of imaging studies are the cornerstone of diagnosis and pre-treatment staging. An abdominal ultrasound is typically the first imaging test performed to confirm a kidney mass. For more detailed evaluation, a computed tomography (CT) scan or a magnetic resonance imaging (MRI) scan is used. These scans provide high-resolution images that help doctors determine the size of the tumor, its involvement with major blood vessels, and whether it has spread to nearby lymph nodes or other organs, such as the lungs and liver. A chest x-ray is also often used to check for lung metastases. Laboratory tests, including blood work and urine analysis, are also performed to screen for tumor-associated conditions like anemia, hypertension, or kidney malfunction.
Biopsy, which involves taking a tissue sample for microscopic examination, is not always performed before surgery. In certain protocols, particularly in North America, a pre-operative biopsy is often avoided to prevent the potential for tumor spill, which could upstage the disease. However, a biopsy may be necessary if imaging indicates the tumor is not a classic Wilms tumor or if the tumor is too large for immediate surgical removal. It is essential to differentiate Wilms tumor from other pediatric renal tumors, such as malignant rhabdoid tumor of the kidney (MRTK), congenital mesoblastic nephroma, and renal cell carcinoma, as these require distinct treatment protocols and have different prognoses.
4.2. Staging and Histology
Once a diagnosis is made, the tumor’s stage and histology are the two most critical factors that guide treatment and determine prognosis.
Tumor Staging: The stage of the tumor describes the extent of its spread. In the United States, the Children’s Oncology Group (COG) staging system is the most commonly used, with staging based on the findings from a post-surgical pathological evaluation.
- Stage I: The tumor is confined to one kidney and was completely removed during surgery
- Stage II: The tumor has grown beyond the kidney but was completely removed, with no visible cancer left behind
- Stage III: Residual cancer remains in the abdomen after surgery
- Stage IV: The cancer has spread to distant organs, such as the lungs or liver
- Stage V: Tumors are present in both kidneys at the time of diagnosis
In Europe, the International Society of Pediatric Oncology (SIOP) protocol is widely used, which stages the tumor based on imaging findings after an initial course of preoperative chemotherapy. The two systems’ differing staging methods are a direct consequence of their distinct clinical philosophies regarding the timing of surgery. While the COG approach provides an immediate, definitive pathological stage, the SIOP approach uses chemotherapy to shrink the tumor first, which can potentially alter the final stage.
Tumor Histology: Histology, or the microscopic appearance of the tumor cells, is a more powerful prognostic indicator than stage alone. The two main histological classifications are:
- Favorable Histology (FH): The most common type, characterized by the classic triphasic pattern and a well-differentiated appearance.
- Anaplastic (Unfavorable) Histology (UH): A less common but more aggressive type, accounting for about 5% of cases. It is defined by the presence of markedly enlarged, hyperchromatic nuclei and multipolar mitotic figures. The presence of anaplasia is a strong predictor of chemoresistance and is associated with a significantly worse prognosis
5. Contemporary Treatment Strategies: The Multimodal Approach
The treatment of Wilms tumor is a highly successful example of modern multimodal therapy, integrating surgery, chemotherapy, and radiation. The choice of treatment is tailored to the individual child and is based primarily on the tumor’s stage and histology.
5.1. A Comparative Analysis of COG and SIOP Protocols
There is a long-standing, fundamental difference in treatment philosophy between the Children’s Oncology Group (COG) in North America and the International Society of Pediatric Oncology (SIOP) in Europe, primarily concerning the timing of surgery.
- COG Protocol: This approach favors primary, upfront surgical removal of the tumor for most cases. The rationale is to obtain immediate, definitive pathological staging, which then dictates the post-operative chemotherapy and radiation regimen. A pre-operative biopsy is typically avoided to reduce the risk of tumor spill
- SIOP Protocol: This approach recommends a course of preoperative chemotherapy for all patients after diagnosis. The goal is to shrink the tumor, making the subsequent surgical resection safer and less extensive. This approach also aims to treat any potential micrometastatic disease early.
While these protocols differ significantly in their sequence of interventions, large-scale clinical trials have demonstrated that the overall survival rates are similarly high, exceeding 90% for both approaches. The similarity in outcomes, despite a profound difference in clinical strategy, highlights the efficacy of a collaborative, protocol-driven approach and the robustness of modern oncology. Both protocols agree on using preoperative chemotherapy for bilateral tumors (Stage V) to allow for nephron-sparing surgery, as preserving kidney function is a paramount concern.
5.2. Surgical Management
Surgical resection is the standard of care for Wilms tumor. The most common procedure is a radical nephrectomy, which involves removing the affected kidney, the adrenal gland on top of it, and surrounding lymph nodes. In the case of bilateral tumors (Stage V), a small biopsy is first taken to confirm the diagnosis, followed by chemotherapy to shrink the tumors. This is done to facilitate nephron-sparing surgery (NSS), a technique that removes only the tumor while preserving healthy kidney tissue. NSS is a significant advancement that reflects a modern paradigm shift in pediatric oncology: as cure rates have improved, the focus has shifted from “cure at all costs” to “cure with minimal long-term consequences”. The success of this technique, which helps children avoid lifelong dialysis or kidney transplantation, has led to its increased use for small, unilateral tumors in selected cases and by experienced surgeons.
5.3. Systemic Therapy and Radiation
Chemotherapy is a critical component of treatment for nearly all cases of Wilms tumor. Standard regimens for favorable histology tumors typically include a combination of vincristine and actinomycin D, with doxorubicin added for more advanced stages. For high-risk tumors, such as those with anaplastic histology, more intensive multi-drug regimens are used, often including drugs like etoposide, cyclophosphamide, and carboplatin.
Radiation therapy is also a cornerstone of treatment for more advanced stages of Wilms tumor (Stage III, IV, and V) and for tumors with anaplastic histology. Modern radiation techniques, such as intensity-modulated radiation therapy (IMRT) and proton beam therapy, use advanced imaging and computing to precisely target the tumor while minimizing exposure to surrounding healthy tissues. This is particularly important for young children, whose developing bodies are highly sensitive to the effects of radiation. Proton beam therapy, for example, is advantageous because protons release most of their energy at a specific depth, causing less damage to tissues located before and after the tumor, in contrast to traditional x-rays. This technological advancement directly addresses the central challenge of long-term side effects by allowing for a more focused delivery of therapeutic radiation.
6. Prognosis and Survival Outcomes
Wilms tumor is widely regarded as one of the most curable pediatric malignancies, a testament to decades of collaborative research and protocol-driven care. The overall 5-year survival rate for children diagnosed with Wilms tumor in the United States is approximately 96%. However, this impressive statistic represents a broad average, and the specific prognosis is highly dependent on key indicators, with tumor histology being the most powerful prognostic factor.
The prognosis for Wilms tumor is directly tied to the stage of the disease at diagnosis and its histological classification. As a general rule, the lower the stage at diagnosis, the better the outcome. Tumors with favorable histology have a significantly better prognosis than those with anaplastic histology, as anaplasia is a strong indicator of chemoresistance and a higher risk of recurrence. A comparison of survival rates by both stage and histology demonstrates that the microscopic characteristics of the tumor can be more indicative of the outcome than the physical extent of its spread. For instance, a child with Stage I favorable histology has a near-perfect prognosis, while a child with Stage III anaplastic histology faces a significantly lower survival rate, highlighting the importance of the tumor’s underlying biology. Age at diagnosis is also a factor, with children under the age of 2 generally having a better prognosis than older children.
Table 2: 4-Year Overall Survival Rates by Stage and Histology
7. Long-Term Survivorship and Late Effects of Treatment
The high cure rates for Wilms tumor have given rise to a new and pressing challenge in pediatric oncology: managing the long-term health consequences of the very treatments that saved a child’s life. The goal of modern care is not just to cure the disease but to do so while minimizing the lifelong morbidity for survivors. These long-term effects can emerge years or even decades after treatment has concluded.
A primary concern is the risk of reduced kidney function or outright kidney failure, as many survivors live with only one kidney. This condition necessitates lifelong monitoring and careful management, including staying well-hydrated and avoiding medications that could strain the remaining kidney. The chemotherapy drug doxorubicin is known to be cardiotoxic, and its use is associated with a higher risk of heart problems later in life, particularly when combined with radiation to the chest. Children treated with this drug require regular heart monitoring with tests like echocardiograms to check for any long-term effects. Radiation therapy can also lead to a range of chronic issues. Radiation to the chest, for instance, can result in long-term lung problems. When applied to the spine, it can cause underdevelopment of soft tissue and skeletal deformities like scoliosis, although modern, more precise techniques have reduced these risks.
A particularly serious late effect is the slightly increased risk of developing a secondary cancer. The most significant risk factors for a second malignancy are prior radiation therapy and the use of certain chemotherapy drugs, notably doxorubicin. For example, young girls who received radiation therapy for lung metastases have an increased risk of breast cancer later in life. Another concern is fertility, as radiation to the abdomen can damage the ovaries in girls, potentially leading to fertility problems or premature menopause. This reality highlights the complex trade-off that clinicians must navigate between providing a curative treatment and ensuring a high quality of life for the survivor. The recognition and management of these late effects are now a central pillar of post-treatment survivorship care.
8. Current Research and Future Directions
The field of Wilms tumor research continues to evolve, building upon its success to refine and personalize treatment strategies. A key focus is the de-escalation of therapy for low-risk patients. Large-scale, collaborative studies by organizations like the Children’s Oncology Group (COG) are investigating whether children with tumors having very favorable features can be cured with less intense treatment, thereby avoiding the long-term side effects associated with more aggressive regimens. For instance, research suggests that some children with lung metastases may not require radiation therapy to the lungs if the metastases respond completely to chemotherapy, a finding that could spare them from potential long-term pulmonary issues.
Genetic and molecular research remains a vital area of study. Scientists are working to identify specific chromosomal and gene changes that predict a tumor’s aggressiveness or its likelihood of relapse. This research provides the foundation for truly individualized, risk-stratified treatment, ensuring that patients who need more intense therapy receive it while those who do not are spared from unnecessary side effects. This knowledge is also driving the development of new, targeted drug therapies. Clinical trials are testing novel agents that can specifically attack the molecular drivers of tumor growth. For example, the drug tegavivint is being evaluated in a Phase I/II trial for recurrent or refractory solid tumors, including Wilms tumor, by targeting the beta-catenin pathway, a key signaling cascade involved in cell proliferation. Other clinical trials are exploring the use of targeted drugs like repotrectinib and cabozantinib-s-malate for tumors with specific gene changes. Immunotherapy, such as B7-H3-specific CAR T-cell therapy, is also being investigated as a potential future treatment modality for solid tumors, which could one day be applied to Wilms tumor.
These research areas are not isolated; they are deeply interconnected. The high survival rates and prevalence of long-term side effects are the primary motivators behind the de-escalation of therapy and the improvement of surgical techniques. The discovery of specific genetic mutations provides the molecular targets for the development of new, less toxic drugs. The existence of a global, collaborative framework, epitomized by the COG and SIOP, is what makes these large-scale clinical trials possible. Together, these efforts represent a concerted, field-wide strategy to perfect the cure for Wilms tumor, moving beyond mere survival to a future where survivors can live long and healthy lives with minimal sequelae from their treatment.
9. Conclusion: Summary and Outlook
Wilms tumor stands as a remarkable success story in the history of pediatric oncology. Significant advancements in multimodal therapy, driven by the collaborative efforts of major international research groups, have transformed a once-fatal diagnosis into a highly curable condition with a near-perfect prognosis for most children. The foundational understanding of the disease, from its developmental origins in nephrogenic rests to the specific genetic mutations that drive its growth, has enabled a sophisticated approach to diagnosis and treatment.
The high cure rates have fundamentally shifted the focus of care toward minimizing the long-term, treatment-related morbidity for survivors. This new challenge is a direct consequence of the field’s success and is driving the next generation of clinical research. The current debate between upfront surgery (COG) and preoperative chemotherapy (SIOP) is a testament to the ongoing effort to refine the therapeutic sequence. The growing adoption of nephron-sparing surgery and minimally invasive techniques further reflects this commitment to preserving long-term quality of life. Looking ahead, the integration of molecular diagnostics into clinical practice and the development of targeted drug therapies hold immense promise for further personalizing care. The ultimate goal is to move toward a future where treatment for Wilms tumor is not only curative but also precise and safe enough to ensure that survivors can lead full and healthy lives free from the burden of chronic health complications.




Leave a Reply